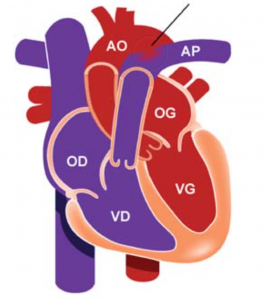
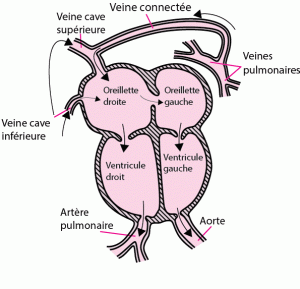
Le cœur est constitué de 4 cavités, deux ventricules et deux oreillettes. Les valves sont comme des portes qui s’ouvrent pour laisser passer le sang, et se referment pour éviter que le sang ne revienne en arrière. Il existes deux valves auriculo-ventriculaires : La valve mitrale entre l’oreillette gauche (OG) et le ventricule gauche (VG), et la valve tricuspide entre l’oreillette droite (OD) et le ventricule droit (VD) ; et deux valves artérielles : La valve aortique entre le VG et l’aorte et la valve pulmonaire entre le VD et l’artère pulmonaire.
Le cœur est constitué de 4 cavités, deux ventricules et deux oreillettes. Les valves sont comme des portes qui s’ouvrent pour laisser passer le sang, et se referment pour éviter que le sang ne revienne en arrière. Il existes deux valves auriculo-ventriculaires : La valve mitrale entre l’oreillette gauche (OG) et le ventricule gauche (VG), et la valve tricuspide entre l’oreillette droite (OD) et le ventricule droit (VD) ; et deux valves artérielles : La valve aortique entre le VG et l’aorte et la valve pulmonaire entre le VD et l’artère pulmonaire.
Le sang riche en oxygène (rouge) proviens des poumons (qui apportent l’oxygène par la respiration) par les veines pulmonaires et poursuit sa route vers les cavités gauches (OG, VG). Le ventricule gauche est une pompe qui va permettre d’éjecter ce sang oxygéné à travers l’Aorte vers les organes (le cerveau, le rein le foie, la peau etc…).
Une fois que les organes se sont servis en oxygène, le sang deviens pauvre en oxygène (bleu) et reviens au cœur par les veines caves vers les cavités droites (OD,VD). Le ventricule droit est également une pompe qui va permettre d’éjecter ce sang pauvre en oxygène à travers l’artère pulmonaire vers les poumons pour se réapprovisionner en oxygène. Et ainsi de suite, le sang rouge retourne au cœur.
C’est donc un circuit en série ou les cavités droites et gauches ont leur propre taches indépendantes et ne communiquent pas entre elles.
 Il s’agit d’une communication anormale entre l’oreillette droite et l’oreillette gauche. Il existe plusieurs type de CIA.
Il s’agit d’une communication anormale entre l’oreillette droite et l’oreillette gauche. Il existe plusieurs type de CIA.
– La CIA ostium secundum au centre de la paroi.
– La CIA ostium primum qui est proche du centre du cœur qui es souvent associée à une malformation appelée canal atrioventriculaire
– La CIA sinus venosus qui est proche des veines caves et qui est souvent associée à une anomalie du retour veineux pulmonaire
– La CIA du sinus coronaire, plus rare, concerne une zone ou se drainent les artère coronaires du cœur.
Seules les CIA ostium secundum peuvent être fermes par cathétérisme cardiaque, les autres nécessitent une chirurgie qui se fait en général vers l’âge de 5 ans.
Le foramen ovale est également une petite communication entre les oreillettes qui est présente pendant la vie fœtale et qui se ferme généralement après la naissance. Dans un nombre non négligeable de cas (environ 1/5 de la population), cette communication ne se ferme pas et on parle de persistance du foramen ovale. La plupart des personnes vivent avec ce FO sans en connaitre l’existence mais dans certain cas, il peut être responsable d’embolies paradoxales gazeuses (plongée sous-marine) ou thrombotiques (caillot sanguin) et provoquer des accidents vasculaires. Dans ces cas, la fermeture de cette communication sera envisagée.
 Il s’agit d’une communication anormale entre les ventricules droits et gauches. Il existe plusieurs types de CIV.
Il s’agit d’une communication anormale entre les ventricules droits et gauches. Il existe plusieurs types de CIV.
– Les CIV musculaires, souvent petites, peuvent se fermer toutes seules en quelque mois.
– Les CIV périmembraneuses, peuvent aussi se fermer toutes seules mais peuvent nécessiter une chirurgie si elles sont larges ou si elle entrainent des difficultés respiratoires ou de prise de poids dans les premiers mois de vie.
– Les CIV de la voie d’ejection (anciennement appelées conoventriculaires) ne se ferment jamais toutes seules. Elles sont sous les gros vaisseaux et sont soit isolées soit associées à d’autres anomalies dans des malformations plus complexes que l’on appelle les cardiopathies conotroncales. Lorsqu’elles sont isolées, la chirurgie est réalisée en général entre 4 et 6mois.
– Les CIV d’admissions, qui sont proches de la croix du cœur (zone centrale ou se trouvent les valves auriculo-ventriculaires) et qui sont souvent associées à une cardiopathie appelée CAV (canal atrioventriculaire). La chirurgie est également prévue en général entre 4 et 6mois.
 Pendant la vie fœtale, le fœtus ne respire pas par les poumons et c’est grâce au placenta que le sang est approvisionné en oxygène.
Pendant la vie fœtale, le fœtus ne respire pas par les poumons et c’est grâce au placenta que le sang est approvisionné en oxygène.
Il existe donc un petit canal, entre l’artère pulmonaire AP et l’aorte, appelé canal artériel qui permet de court circuiter les poumons avant la naissance.
Ce canal se ferme normalement dans les premiers jours de vie.
Parfois, ce canal ne se ferme pas et reste ouvert et persistant après la naissance.
S’il ne gêne pas le nouveau-né, on peut patienter jusqu’à ce qu’il se ferme spontanément.
S’il est large et s’il induit des difficultés alimentaires et respiratoires, il nécessite une fermeture qui peut se faire soit par cathétérisme cardiaque soit par chirurgie.
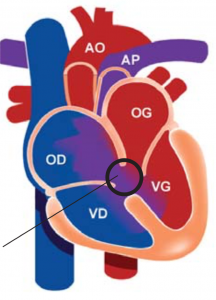 Le CAV complet est une malformation cardiaque qui associe plusieurs anomalies au niveau de la croix du cœur (zone centrale) :
Le CAV complet est une malformation cardiaque qui associe plusieurs anomalies au niveau de la croix du cœur (zone centrale) :
– Une CIA ostium primum (voir CIA)
– Une CIV d’admission (voir CIV)
– Une seule valve auriculo-ventriculaire entre les oreillettes et les ventricules au lieu de deux.
Parfois ces associations sont incomplètes comme dans le CAV partiel où la CIV est absente ou le CAV intermédiaire où la CIV est petite.
Cette malformation est caractéristique de la Trisomie 21.
Pour le CAV complet, la chirurgie doit être réalisée être réalisée entre 4 et 6mois de vie.
Pour le CAV partiel, la chirurgie est réalisée en général, comme pour les CIA vers l’âge de 5ans, ou plus tôt en cas de complications.
Pour le CAV intermédiaire, le timing de la chirurgie dépendra de la tolérance clinique.
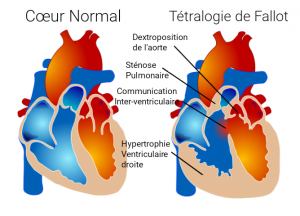 C’est une malformation qui associe plusieurs anomalies :
C’est une malformation qui associe plusieurs anomalies :
– Une CIV de la voie d’ejection (voir CIV) avec déplacement de l’aorte vers la droite (dextroposition).
– Un obstacle (sténose) au niveau de la valve pulmonaire et la zone d’éjection sous la valve pulmonaire, créant une hypertrophie (muscularisation) du ventricule droit.
Cette malformation peut être associée à des maladies génétiques comme la trisomie 21 par exemple.
En fonction de l’obstacle pulmonaire, parfois les enfants peuvent être cyanosés (bleus) à la naissance.
Lorsque le diagnostic est fait à la naissance, on prévoit une chirurgie en général vers les 6 mois de l’enfant.
Si l’obstacle est très serré, et que l’enfant est très bleu ou fait des malaises, on a parfois recourt à une chirurgie intermédiaire avant la cure complète.
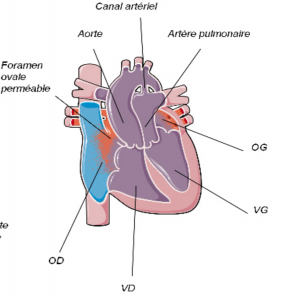 C’est une malformation ou les gros vaisseaux (l’artère pulmonaire et l’aorte), sont inversés. Dans ce cas, l’aorte sort du ventricule droit et l’artère pulmonaire du ventricule gauche. Les organes sont mal oxygénés car du sang bleu est éjecté dans l’aorte. Les nouveau-nés sont donc cyanosés (bleus) à la naissance.
C’est une malformation ou les gros vaisseaux (l’artère pulmonaire et l’aorte), sont inversés. Dans ce cas, l’aorte sort du ventricule droit et l’artère pulmonaire du ventricule gauche. Les organes sont mal oxygénés car du sang bleu est éjecté dans l’aorte. Les nouveau-nés sont donc cyanosés (bleus) à la naissance.
En attendant la chirurgie qui a lieu en général dans la première semaine de vie, il est possible d’améliorer l’oxygénation des organe en ouvrant des communications entre les deux circulations :
– Le canal artériel peut être maintenu ouvert grâce à un médicament injecté par voie veineuse : les prostaglandines (non disponible à la commercialisation au Maroc).
– Le foramen ovale peut être élargit grâce à une manœuvre réalisée par cathétérisme cardiaque (une sonde est montée dans le cœur par les veines) : la manœuvre de Rashkind.
Le traitement chirurgical consiste en un rétablissement des connexions normales en ré-inversant les vaisseaux: le switch artériel.
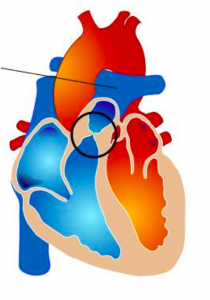 Il s’agit d’un rétrécissement d’une des valves qui sépare les ventricules des gros vaisseaux. Il existe dans les deux cas des formes néonatales qui doivent être prises en charge souvent en urgence, et des formes de l’enfant plus grand ou de l’adulte.
Il s’agit d’un rétrécissement d’une des valves qui sépare les ventricules des gros vaisseaux. Il existe dans les deux cas des formes néonatales qui doivent être prises en charge souvent en urgence, et des formes de l’enfant plus grand ou de l’adulte.
Le traitement sera réalisé soit par cathétérisme soit par chirurgie selon le type de pathologie et le contexte.
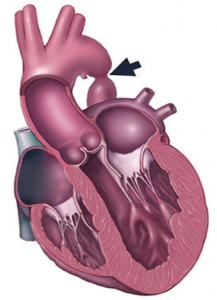 Cette malformation correspond à un rétrécissement de l’aorte au niveau de son isthme. Il existe deux formes :
Cette malformation correspond à un rétrécissement de l’aorte au niveau de son isthme. Il existe deux formes :
– La forme néonatale qui survient dans les premiers jours ou semaines de vie lorsque le canal artériel se ferme et qui peut engendrer une insuffisance cardiaque aigüe et mettre en péril le nouveau-né.
– La forme de l’adulte qui est d’évolution lente et qui est souvent diagnostiquée sur une hypertension artérielle.
Le traitement de la forme néonatal est chirurgical et consiste à réparer la zone rétrécie. Dans certain cas, si la situation est critique, il est possible de recourir à une dilatation par cathétérisme cardiaque (à travers les veines).
Le traitement de la forme adulte peut être chirurgical ou par cathétérisme (avec mise en place d’un stent) selon les cas.
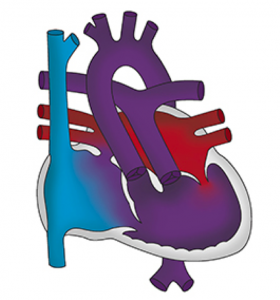 Il existe plusieurs types de malformations que l’on regroupe sous le terme « ventricule unique », ce qui sous-entend que le cœur ne fonctionne qu’avec un seul ventricule. Il peut s’agir d’une hypoplasie (défaut de développement) du ventricule gauche, ou du ventricule droit ou alors même d’une malformation, qui pour une raison ou pour une autre ne peut être réparée en conservant les deux ventricules.
Il existe plusieurs types de malformations que l’on regroupe sous le terme « ventricule unique », ce qui sous-entend que le cœur ne fonctionne qu’avec un seul ventricule. Il peut s’agir d’une hypoplasie (défaut de développement) du ventricule gauche, ou du ventricule droit ou alors même d’une malformation, qui pour une raison ou pour une autre ne peut être réparée en conservant les deux ventricules.
Ce sont des malformations graves et non curables. Les traitements proposés sont palliatifs, c’est-à-dire qu’ils ne permettront jamais de « réparer » le cœur. Il s’agit d’un traitement séquentiel en 2 ou 3 chirurgies (ou plus) pour aboutir au final à une dérivation cavo-pulmonaire totale qui va dériver le sang oxygéné directement vers le poumon par un tube, et permettre d’utiliser le seul ventricule fonctionnel comme pompe.
Le pronostic de ces malformations est aléatoire et peut être assombri par de nombreuses complications de cette circulation artificielle.
 Il s’agit d’une malformation ou les veines qui doivent amener le sang oxygéné du poumon vers l’oreillette gauche ne s’abouchent pas au bon endroit (dans les veines caves ou dans l’oreillette droite).
Il s’agit d’une malformation ou les veines qui doivent amener le sang oxygéné du poumon vers l’oreillette gauche ne s’abouchent pas au bon endroit (dans les veines caves ou dans l’oreillette droite).
Le RVPA est total si les 4 veines ne s’abouchent pas au bon endroit. Il s’agit d’une malformation qui entraine une grande détresse chez le nouveau-né surtout si l’abouchement de ces veines est bloqué. C’est une urgence chirurgicale.
Le RVPA peut être partiel si seulement une ou deux ou 3 veines ne s’abouchent pas correctement. Dans ce cas, la malformation est souvent dépistée beaucoup plus tard et nécessite une correction chirurgicale sans urgence.
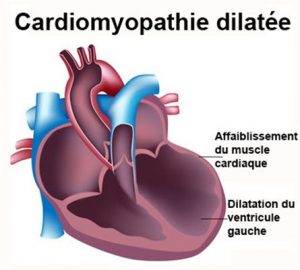 L’insuffisance cardiaque est une situation ou le cœur n’arrive plus à maintenir un débit suffisant pour vasculariser les organes.
L’insuffisance cardiaque est une situation ou le cœur n’arrive plus à maintenir un débit suffisant pour vasculariser les organes.
Cette situation peut survenir de façon aigüe et engendrer une grande détresse et nécessiter de la réanimation, ou de façon chronique ou elle sera découverte sur des signes cliniques caractéristiques (comme un retard de croissance, un essoufflement lors des efforts, des douleurs abdominales).
Les causes de l’insuffisance cardiaque sont multiples, il peut s’agir de:
– Malformations cardiaques : par une surcharge en pression ou en volume, un défaut de perfusion du myocarde (anomalies coronaires)
– Causes toxiques comme certaines chimiothérapies
– Causes infectieuses souvent virales qui entrainent des myocardites
– Troubles du rythmes ou de conduction entrainant une défaillance cardiaque
– Maladies métaboliques souvent congénitales ou le cœur est une atteinte parmi d’autres organes atteints (le cerveau le foie)
– Maladies génétiques héréditaires
– Maladies musculaires comme les myopathies qui affectent le muscle du cœur
Il existe des traitements médicamenteux pour la phase aiguë en réanimation, pour la phase chronique. Mais lorsque la situation est critique, parfois la seule solution est la transplantation cardiaque (qui n’est pas réalisée au Maroc).
 Les tumeurs cardiaques de l’enfant sont rares et souvent bénignes.
Les tumeurs cardiaques de l’enfant sont rares et souvent bénignes.
La tumeur la plus fréquente est le rhabdomyome, qui est une tumeur souvent multiple, qui a le potentiel de régresser spontanément et disparaitre au bout de quelque mois. Certaines situations nécessitent parfois une résection chirurgicale si la tumeur gène le fonctionnement du cœur. Elle rentre souvent dans le cadre d’une maladie appelée la sclérose tubéreuse de Bourneville.
Les autres tumeurs (tératomes, fibromes, myxomes…) sont encore plus rares et nécessitent souvent une résection chirurgicale.
 Le cœur est doté d’un circuit électrique qui permet de donner des impulsions nécessaires à la contraction synchronisée des oreillettes et des ventricules.
Le cœur est doté d’un circuit électrique qui permet de donner des impulsions nécessaires à la contraction synchronisée des oreillettes et des ventricules.
Parfois, ce circuit est défaillant et peut entrainer :
– Des troubles du rythme auriculaires ou ventriculaires qui peuvent entrainer des tachycardies (accélération du rythme cardiaque). Les traitements sont souvent médicamenteux. Dans de rares cas, il est nécessaire d’ablater (bruler) la zone défectueuse au cathétérisme cardiaque. Certains troubles du rythme ventriculaires nécessitent la mise en place d’un défibrillateur.
– Des troubles de conduction qui entrainent parfois des bradycardies (ralentissement du rythme cardiaque). Les traitements peuvent être médicamenteux dans certaines maladies héréditaires, mais la plupart du temps, le traitement consiste à implanter un pacemaker pour pallier à la défaillance de la pile cardiaque.
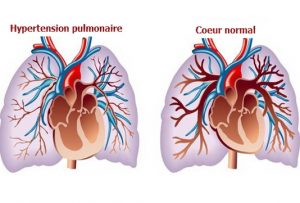
L’hypertension pulmonaire est une situation ou la pression dans les poumons est augmentée alors que c’est un circuit qui fonctionne normalement à faible pression. Cela a pour conséquence d’affaiblir le ventricule droit qui “lutte” contre cet obstacle.
Cette hypertension peut être:
- Primitive, l’hypertension artérielle pulmonaire est une maladie ou les artères et artérioles pulmonaires sont trop « musclées ». De nombreux gènes ont été découverts ces dernières années. C’est une maladie grave avec un pronostic sombre. Les traitements ne guérissent pas la maladie mais peuvent en ralentir l’évolution. Il s’agit de traitement médicamenteux oraux ou injectés par une pompe. Lorsque la situation est critique, certaine opérations par cathétérisme ou chirurgie peuvent soulager l’hypertension.
- Chez l’enfant il s’agit souvent de malformations cardiaque qui n’ont pas été opérées à temps. Lorsqu’il existe des communications anormales entre les cavités gauches et droites, le shunt peut abimer les artérioles pulmonaires à long terme en entrainer une hypertension pulmonaire. Pour éviter ces situations, le diagnostic précoce des malformations et le respect des délais chirurgicaux est primordial.